USP1通过去泛素化Akt抑制长期饥饿时肌肉中的PI3K-Akt-Foxo信号转导
发布时间:2022-02-18 [返回]
写在前面
今天推荐的是由以色列海法理工学院生物系团队在2020年4月3日发表于EMBO Reports(IF:8.210,JCR 1区)的一篇文章,通讯作者是Shenhav Cohen教授,研究表明长时间饥饿状态下,USP1通过介导Akt的去泛素化抑制PI3K-Akt级联信号转导。
研究背景
PI3K-Akt-FoxO-mTOR信号通路是控制所有细胞生长和代谢的中枢通路。IGF-I或胰岛素激活该通路可促进细胞分裂,相反,抑制这一途径会降低细胞存活率并导致肌肉萎缩。PI3K-Akt信号转导的核心是丝氨酸/苏氨酸激酶Akt,它是细胞表面受体传递生长和生存信号不可或缺的通道。Akt的失调会导致多种疾病,包括癌症、胰岛素抵抗和肌肉萎缩等。Akt的活性在细胞中受到严格调控,PDK-1和mTORC2激酶分别在T308和S473上磷酸化Akt。Akt在这些残基上的磷酸化被认为是限速的,也是最大程度激活Akt的必要条件。另一方面,Akt磷酸化之前的泛素化调控也是PI3K-Akt活性所必需的。
USP1是半胱氨酸蛋白酶USP家族的一员,它通过去泛素化作用来逆转蛋白修饰。USP1最具特色的功能是在细胞核中作为DNA损伤应答的调节因子,并作为某些转录因子的负调节因子阻止细胞分化。最近的研究表明,USP1还能通过促进胰腺β细胞的凋亡在糖尿病中起作用。此前发现一种USP1基因敲除的小鼠有多种发育缺陷,包括骨质减少、围产期致死、雄性不育、染色体不稳定和Fanconi贫血。虽然USP1在DNA修复机制中的重要作用已被充分证明,但其在调节代谢和生长方面的确切功能尚不清楚。
摘要部分
研究发现,去泛素化酶USP1可以去除Akt激酶上K63连接的多泛素链,从而抑制长时间饥饿时小鼠肌肉中的PI3K-Akt-FoxO信号。作者通过DUB筛选平台确定了USP1是Akt的直接DUB。研究表明,小鼠肌肉中USP1的缺失促进了禁食期间Akt的泛素化、PI3K-Akt-FoxO信号转导以及葡萄糖摄取。作者通过免疫共沉淀和质谱鉴定出Dab2、TSC1/TSC2和PHLPP1为USP1的结合蛋白。饥饿状态下,Dab2对于USP1-TSC1-PHLPP1复合物募集Akt以及抑制PI3K-Akt-FoxO信号至关重要。此外,USP1通过限制TSC1水平以维持mTOR介导的基础蛋白合成速率,从而维持其自身的蛋白水平。作者认为,当细胞能量水平较低时,Dab2可以招募Akt到USP1-TSC1-PHLPP1复合物中,从而有效终止生长信号的传递。
研究内容
1. USP1是Akt的去泛素化酶
作者通过免疫印迹法对喂食和禁食(2d)小鼠的骨骼肌提取物进行分析,结果发现Akt在正常条件下处于泛素化状态,但在禁食期间其泛素化水平降低,而未经修饰的Akt累积。这表明,Akt在禁食期间被去泛素化。
接着作者从正常肌肉匀浆中免疫沉淀Akt,并将沉淀加入含有活性DUB阵列的DUB筛选板。结果显示,USP1/UAF1、USP11、OTUB2、OTUD5(p177S)和OTUD6A这五个DUBs切断了Akt上的多泛素链并降低其泛素化水平。其中,USP1优先去除K63连接的泛素链,这是Akt激活所必需的多泛素化类型。
为探究USP1是否能在体内将Akt去泛素化,作者分析了禁食期间USP1敲低对Akt泛素化的影响。结果显示,USP1的下调抑制了Akt泛素化水平的降低,相反,Akt以泛素化形式累积。当作者将USP1(C90S)质粒导入细胞来抑制USP1功能时,也得到了类似的结果。进一步的研究表明,K63泛素化的Akt可能在禁食期间增加,而USP1催化该蛋白的去泛素化。
为确定USP1是否切割Akt的K8-泛素链,作者将shLacz(对照)或HA-Akt(K8R)质粒与USP1(C90S)共转导入肌肉中以引起泛素化Akt的积聚。结果发现,禁食2d时,HA-Akt(K8R)显示出有限的泛素化,远低于仅表达USP1(C90S)的内源性Akt的泛素化水平。这些表明,在禁食期间抑制USP1会使得Akt在K8上发生泛素化。
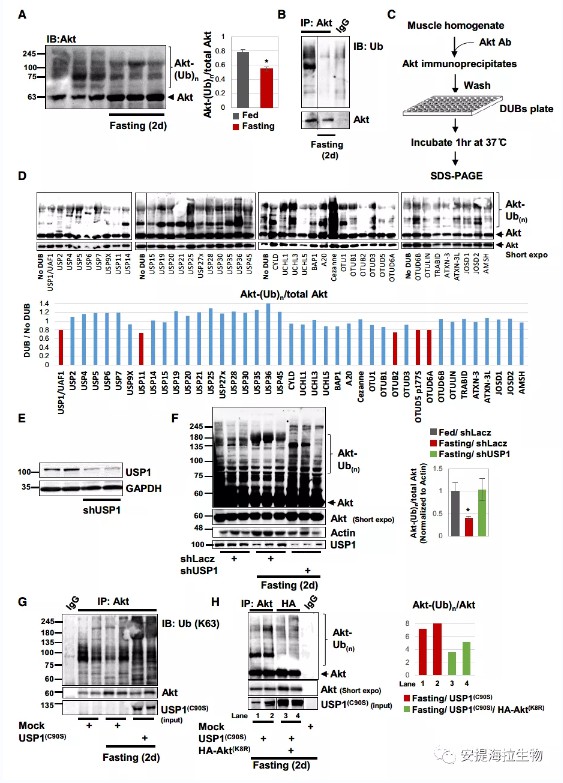
图1. USP1是体内作用于Akt的去泛素化酶
2. USP1介导的去泛素化抑制了Akt在T308的磷酸化以及PI3K-Akt-Foxo信号转导
作者发现,小鼠禁食2天后,胰岛素受体、Akt及其靶点FoxO3和GSK3b的磷酸化水平显著降低。然而,抑制USP1几乎完全阻断了这种现象。事实上,Akt(T308)、FoxO3和GSK3b的磷酸化水平在抑制USP1后与喂食小鼠相似。此外,尽管Akt的泛素化可以促进其在T308和S473的磷酸化,但在USP1被抑制而泛素化的Akt累积的肌肉中,Akt在S473的磷酸化水平仍然较低。
通常在禁食期间,FoxO被激活并刺激萎缩基因的表达,包括MuRF1和Atrogin 1。作者发现,通过shRNA(shUSP1)下调USP1会导致MuRF1和Atrogin1表达显著降低。实验表明,相对于喂食小鼠,禁食小鼠的平均重量减少了31%,但USP1的下调明显减轻了这种消耗。另外,通过注射ML323特异性抑制USP1可显著改善小鼠的糖耐量,并在禁食期间激活PI3K-Akt-FoxO信号通路。
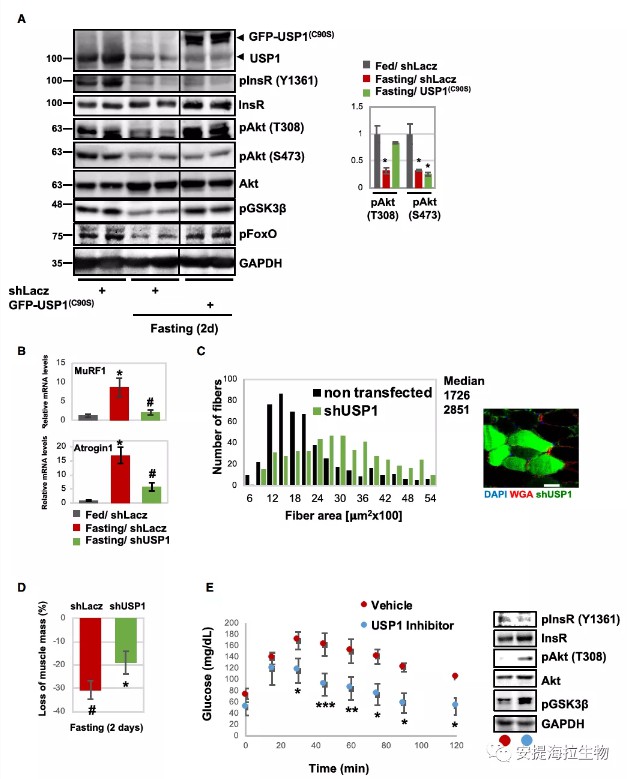
图2. USP1对Akt的去泛素化使其在T308的磷酸化和PI3K-Akt-FoxO信号转导降低
3. Ser/Thr磷酸酶PHLPP1在禁食期间抑制Akt在S473的磷酸化
先前的研究表明,Ser/Thr蛋白磷酸酶PHLPP1能使Akt在S473发生去磷酸化。作者将特定的shRNA(shPHLPP1)导入小鼠TA肌肉来下调PHLPP1水平。结果发现,PHLPP1的降低导致磷酸化的Akt(S473)和S6K(T389)增加,而磷酸化的Akt(T308)保持在较低水平。与Akt和S6K的磷酸化增加一致,PHLPP1下调也促进了蛋白质合成。这种蛋白质合成的增强可以解释在表达shPHLPP1的纤维中观察到的肌肉萎缩被显著抑制的现象。作者还在喂食小鼠的肌肉中发现,PHLPP1和UAF1可与USP1共沉淀。
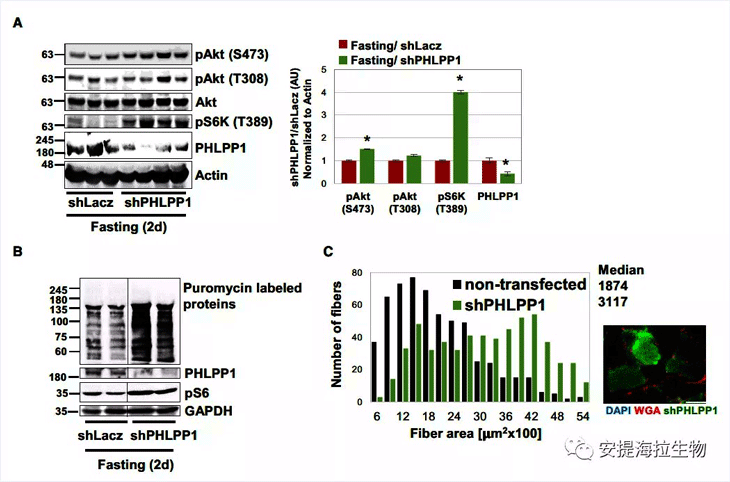
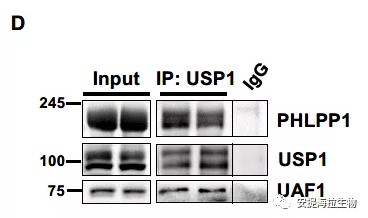
图3. 磷酸酶PHLPP1在禁食时减少Akt在S473的磷酸化
研究结论:禁食期间,PHLPP1介导Akt在S473去磷酸化,其可能与USP1共同发挥作用,从而有效抑制Akt和PI3K-Akt-mTOR信号传导。
4. 禁食时Akt被招募至USP1-PHLPP1复合物中
作者通过免疫沉淀和质谱研究与Akt相互作用的组分,即PHLPP1、USP1及其辅助因子UAF1和FoxO3。质谱分析显示,只有29种蛋白是这四个组分的共同结合物,包括TSC1/TSC2复合物以及可抑制上皮细胞增殖的Dab2。
喂食小鼠肌肉匀浆的甘油梯度分级表明,TSC1、TSC2、USP1/UAF1和PHLPP1沉淀到相同组分中,而Akt在这些组分中几乎不存在。进一步研究表明,在禁食期间,Akt转移到含有USP1/UAF1、TSC1/TSC2和PHLPP1的较重组分中,此时的Akt在T308的磷酸化水平降低。
免疫沉淀实验显示,TSC2、USP1和PHLPP1可与TSC1共沉淀。研究表明,在禁食期间,USP1通过结合UAF1而被激活,并且招募Akt。然而,作者发现USP1与UAF1的结合并不是招募Akt所必需的,因为即使USP1/UAF1的结合受到干扰,Akt仍然可与USP1结合。
作者发现,在禁食期间,当Akt被抑制时,USP1通过影响TSC1提高蛋白质合成速率。TSC1在USP1被抑制时的积累并不是由于其基因表达增加,研究表明,USP1通过促进TSC1的降解来限制其含量。此外作者发现,在饥饿和非饥饿肌管中,USP1的下调均能导致泛素化TSC1的积累、Akt磷酸化水平升高以及S6K磷酸化水平降低。这表明正常情况下,USP1也调节PI3K-Akt信号和TSC1的稳定性。
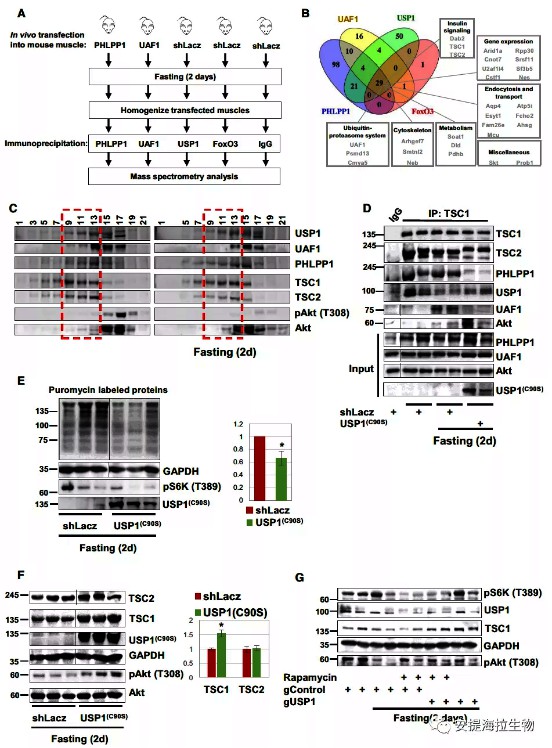
图4. Akt在禁食时被招募到USP1-PHLPP1-TSC1复合物中
研究结论:当PI3K-Akt信号强度较低时,USP1通过限制TSC1以促进蛋白质合成速率,并维持自身蛋白水平。USP1的下调会导致TSC1累积,并且USP1对TSC1的作用与其抑制Akt活性的作用无关。
5. Dab2促进USP1-PHLPP1-TSC1复合物招募Akt
为探究Dab2是否影响禁食期间的PI3K-Akt信号活性,作者使用Dab2-DN载体作为Dab2-Akt结合的抑制剂。实验结果显示,在禁食期间,Akt与USP1、TSC1的结合增强。类似地,Dab2与该蛋白复合体的结合也在禁食时增加。研究发现,抑制Dab2-Akt的结合可以阻止Akt和Dab2被募集到USP1复合物中,这表明,Dab2在其中起作用并促进USP1招募Akt。
为了确定Dab2是否促进Akt去泛素化,作者在喂食和禁食小鼠的肌肉中免疫沉淀Akt,并通过SDS–PAGE和免疫印迹分析。结果显示,在禁食状态下,Akt的高分子量泛素化形态减少。而Dab2功能的抑制导致高分子量K63连接的泛素结合物在Akt上累积。因此,Akt的去泛素化依赖于Dab2的功能。
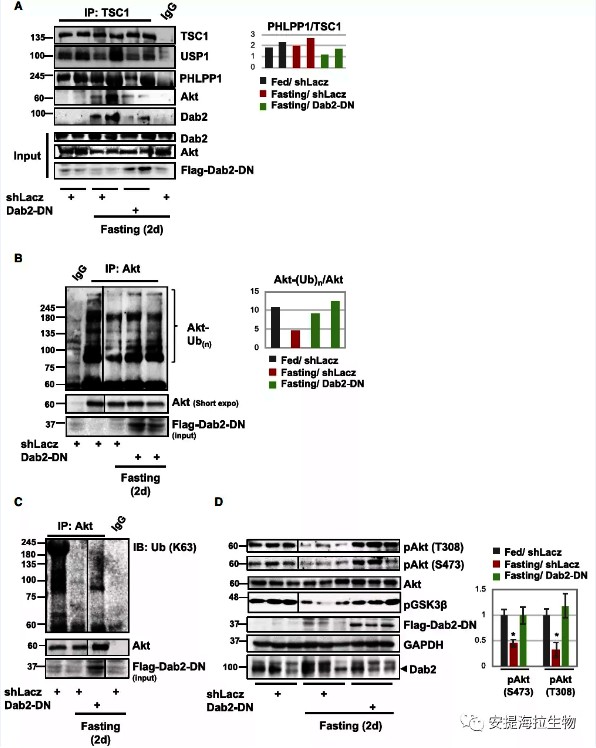
图5. Dab2促进Akt被招募到USP1-PHLPP1-TSC1复合物中
研究结论:Dab2对于禁食状态下USP1诱导的Akt泛素化的减少以及由此产生的Akt和PI3K-Akt-FoxO信号的抑制至关重要。
结论与讨论

Thank you!










Copyright © 厦门安提海拉生物科技有限公司 版权所有 All Rights Reserved.
手机:400-878-2661 邮箱:service@antihela.com 联系人:王先生
技术支持:厦门易尔通网络科技有限公司 闽ICP备15018136号-1 公安备案:
手机:400-878-2661 邮箱:service@antihela.com 联系人:王先生
技术支持:厦门易尔通网络科技有限公司 闽ICP备15018136号-1 公安备案:
